Scientific discovery by video game players
The ability to determine the three dimensional structure of disease-related proteins is essential to our understanding of how they work and how they can be successfully targeted with drugs, yet the gap between the number of known protein sequences and the number of solved protein structures is growing every year. The unusual approach of enlisting online gamers to tackle this problem has shown promise with the protein-folding videogame Foldit. By leveraging human puzzle solving, pattern-recognition, and 3D spatial reasoning, Foldit players are able to outperform many state of the art protein structure prediction methods.
Foldit players have been most successful in the refinement of partially correct models. This field of structure prediction has shown promise in solving crystal structures by molecular replacement (MR), and purely computational methods for this purpose have already proved successful. In one example where state-of-the-art methods were unable to produce a suitable model, Foldit players created models of sufficient quality for successful MR and subsequent structure determination of a monomeric retroviral protease.
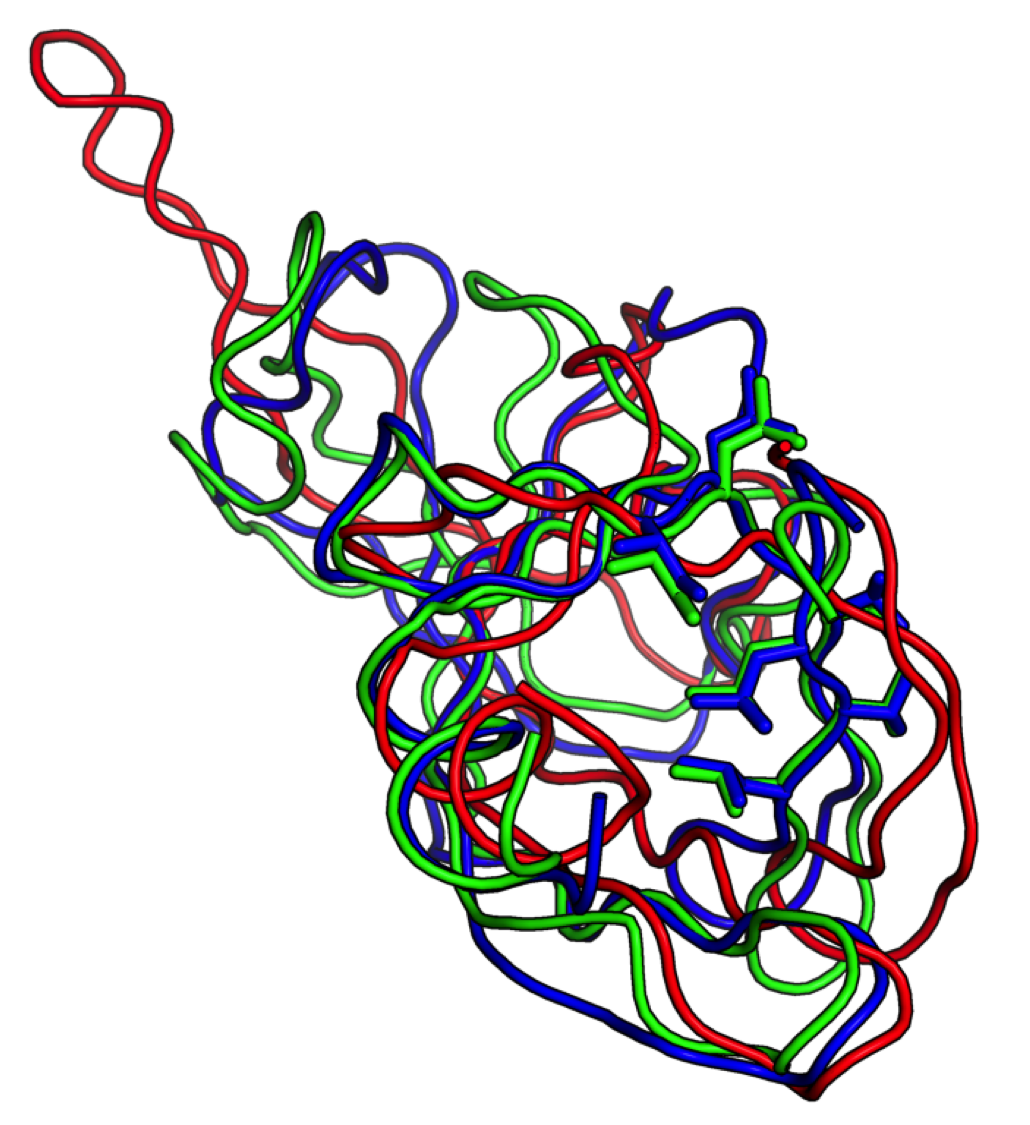
Starting from a low-resolution NMR model (red), Foldit players generated a model (green) of sufficient accuracy to provide an unambiguous molecular replacement solution, allowing rapid determination of the ultimate crystal structure (blue). Researchers had been unable to solve this structure by molecular replacement for over a decade. Foldit players managed to produce this model in ten days without any experimental data.
Our current research focuses on providing Foldit players with experimental data that automated methods currently cannot solve. If you are an experimentalist with electron density data for a structure you have been unable to solve, please contact us (using the Contact Information tab at the top).